* 본 글은 FDA 의 Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions 가이던스를 참고하여 작성되었습니다.
(* This post is written with reference to the FDA's Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions guidance.)
- 가이던스 개요
2023년 4월, FDA 가 인공지능 의료기기와 관련된 새로운 가이드라인을 발표했습니다.
가이던스의 이름은 다음과 같습니다.
Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions (인공 지능/머신 러닝 소프트웨어를 위한 사전 결정된 변경 제어 계획에 대한 마케팅 제출 권장 사항)
가이던스 내에서는 PCCP 라고도 부르고 있으며, PCCP란 Predetermined Change Control Plan 으로, 사전정의된 변경 계획 정도로 번역하면 될 것 같습니다.
PCCP 가이던스 문서의 목차는 다음과 같습니다.
- PCCP 를 제작하게 된 배경
- PCCP 및 관련 정의
- PCCP 정책 및 해설
- 변경사항에 대한 설명 및 변경 절차
- (PCCP로 인한) 향후 영향
인공지능 의료기기, 즉 AI/ML Medical Devices 들은 소프트웨어 제품으로서만이 아닌, 주요 기능인 인공지능 모델에 대한 잦은 업데이트를 필요로 합니다. FDA 는 인공지능에 관련된 여러 가이드던스에서 관련된 내용을 고민해왔습니다. FDA는 2019년 4월에 발표되었던 Artificial Intelligence and Machine Learning in Software as a Medical Device 을 시작으로, Deciding When to Submit a 510(k) for a Software Change to an Existing Device 와 같은 가이던스에서 SaMD의 업데이트와 관련된 가이던스를 발표했습니다.
다만 위 가이던스들로는 인공지능 모델의 업데이트에 대해 별도 안내를 주기는 어려웠습니다. 아마도 현업 종사자들 또한 많은 고민을 진행했을 것 같습니다. 모델의 업데이트는 기기의 성능을 더욱 높일 수 있을 뿐만이 아니라, 학습 데이터가 변경되었다는 것은 이미 해당 기기가 더 이상 같은 제품으로서 취급할 수 없다고 판단되었을 수도 있습니다.
2019년 가이던스에서 이미 해당 부분을 고민한 내용이 있습니다. (page 3)
"What we heard: Stakeholders provided many suggestions for further development of the proposed regulatory framework for AI/ML-based SaMD, including for the Predetermined Change Control Plan
described in the discussion paper. What we’ll do: Update the proposed framework for AI/ML-based SaMD, including through issuance of Draft Guidance on the Predetermined Change Control Plan "
- (이해관계자들로부터 AI/ML 기반 SaMD 에 대한 제안된 규제 프레임워크의 추가 개발 제안이 요청됨)
- (AI/ML 기반 SaMD 에 대한 프레임워크 업데이트 진행)
여기서 Predetermined Change Control Plan 이 등장합니다. SaMD는 SPS(SaMD Pre-Specficiations, SW의료기기 사전명세)에 따라 제품모델의 학습에 따른 사전정의를 내릴 수 있고, APC(Algorithm Change Protocol, 알고리즘 변경 절차)에 따라 모델 알고리즘이 어떤 방식으로 업데이트가 이루어질 것인지 절차를 구성할 수 있습니다. 2019년 해당 가이던스에서는 이와 관련하여, SPS와 APC 외에도 여러 PCCP 에 대한 제안들이 있다고 추가 언급하고 있습니다. 이에 대해서는 본 2023년 가이던스에서는 "Modification Protocol" 을 언급하며 구체적으로 제시합니다. 또한 본 가이던스에서는 ML-DSF 라는 단어와 함께 AI 모델 업데이트를 이야기합니다. ML-DSF 란 Machine Learning-Enabled Device Software Function 을 말합니다.
(개인 의견) FDA 가 굳이 PCCP 에 대한 가이던스를 발표한 이유를 짐작해보자면, 많은 양의 510(k) Submission 이 반복되어 제출되기 때문일 것으로 보입니다. 이와 관련해서는 다음과 같은 언급이 있습니다.
"By including a PCCP in a marketing submission, manufacturers can proactively pre-specify and seek premarket authorization for intended modifications (and their method of implementation) to an ML-DSF without necessitating additional marketing submissions for each modification delineated and implemented in accordance with the PCCP. In other words, a PCCP, as part of a marketing submission, is intended to provide a means to implement modifications to an ML-DSF that generally would otherwise require additional marketing submissions prior to implementation."
물론, PCCP 에 포함되지 않는 경우 사전 승인이 필요하다고 언급하고 있습니다. (line 180-191, page 6)
그렇다면 구체적으로 PCCP 가 어떤 절차와 경우를 포함하고 있는지 알아보겠습니다.
- PCCP 개요
가이던스의 다섯번째 Section 부터 PCCP 에 대한 구체적인 내용을 제공합니다.
V. Policy for Predetermined Change Control Plans (V. 사전 결정된 변경 관리 계획에 대한 정책) 중 A ~ E 를 보면 PCCP 의 절차에 대해 언급합니다.
A: Components of a PCCP : Sections VI. – VIII 를 참고하여 진행, PCCP 구성 요소들은 서로 연계되어야 함
B: Establishing a PCCP : PCCP 가 있는 경우 510(k), PMA, De Novo 모두 제출 시 고려하여 설정되어야 함. 단,
- 510(k) 경우 PCCP 가 포함된 predicate device 와 SE를 진행할 시, 버전을 명시할 것.
- PCCP 를 포함한 기 허가된 제품에 새로운 PCCP 를 추가할 경우, 적절한 제출 요건을 포함할 것. (기 허가된 제품에서 주요 변경사항 없이 PCCP 만 추가할 경우, FDA 는 PCCP 에 집중 리뷰할 예정)
- PCCP 에 대한 의견을 받을 수 있도록 Q-Submission 절차를 활용 권고
- Pre-Submission 에서 PCCP 내용 승인을 진행하진 않음
C: Identifying a PCCP in a Marketing Submission : PCCP 는 submission 에서 독립 섹션으로 포함되어야 함.
- Coverletter 에서 눈에 띄도록 언급, 포함되어야 함
- submission 목차에 ""Predetermined Change Control Plan" 항목을 언급, 포함되어야 함.
- PCCP는 device description, labeling, 그리고 연관 섹션(SE 및 safety and effectiveness)에서 언급되어야 함.
- device labeling 의 경우, 적절한 사용 방법(adequate directions for use) 을 포함하여야 하며, change control plan 이 (계획에 따라 장치가 변경될 때 안전하고 효과적으로 사용될 수 있도록) labeling을 포함하도록 권고할 수 있음
- PCCP 는 510(k) summary, De novo decision summary, PMA SEED summary 에서 설명되어야 함.
추가적으로, C section 에서는 다음 부분이 중요해 보입니다.
For ML-SFs with an authorized PCCP, the labeling should explain that the device incorporates machine learning and has a PCCP so that users are aware that the device may require the user to perform software updates, and that such software updates may modify the device’s performance, inputs, or use.
PCCP 가 승인된 ML 제품의 경우, 라벨링에서 해당 장치가 머신러닝을 포함하고 있으며, 사용자가 업데이트를 수행할 시 이로 인해 장치의 성능, 입력, 사용이 바뀔 수 있음을 설명해야 함. (should explain)
D. Utilizing an Authorized PCCP to Implement Device Modifications : PCCP 가 submission 에서 검토되면, PCCP 가 승인의 일부로 간주됨. (따라서 승인받은) PCCP 포함 제품은 업데이트가 PCCP 와 일치할 경우 신규 submission 이 필요하지 않음. 단, 제조업체는 이 사항을 자사의 품질 시스템에 따라 기록.
- 업데이트가 PCCP 내용에 해당하는지의 여부는, 다음 그림에서 절차가 언급됩니다.

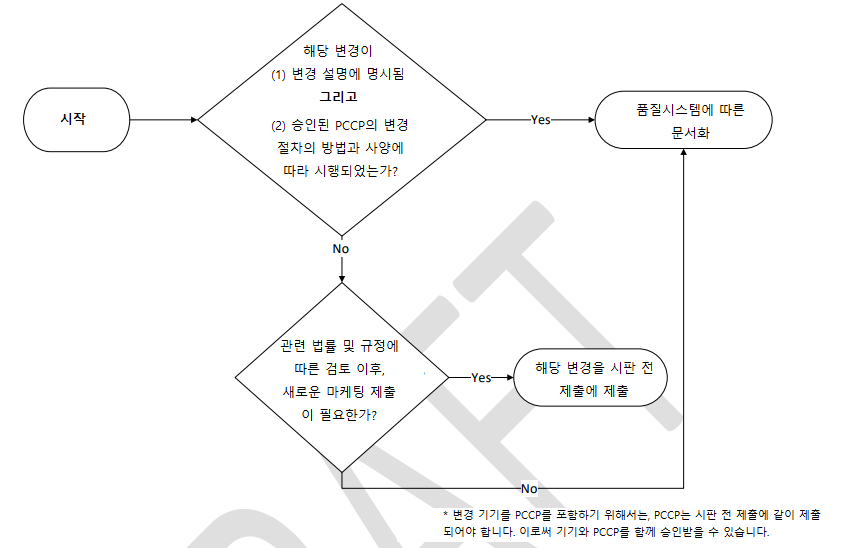
(* 보시면 아시겠지만 안타깝게도 위 Figure 만으로는 PCCP 여부 확인 절차가 충분히 설명되진 않고 있습니다)
E. Modifying a PCCP for an Authorized Device : 기 승인된 장치에 대한 PCCP 변경은 신규 submission 진행
- 기기에 PCCP 포함 변경은 PMA, De Novo, 510(k) 신규로 진행될 것으로 보임
- 이는 FDA는 PCCP 변경 부분이 기기의 안정성 유효성에 상당한 영향을 미칠 것으로 예상하기 때문
- 기 승인된 제품은 PCCP 변경 submission 을 진행할 경우, FDA 는 PCCP 에 집중적으로 검토할 예정
- Modification 개요
그렇다면, 본 가이던스에서 말하는 변경(modification)란 무엇인지, 어디까지가 PCCP 에서 말하는(혹은 허용하는) 변경인지가 궁금해집니다. 이는 VI. Description of Modifications 에서 언급하고 있습니다.
...To ensure an efficient review, FDA recommends that a PCCP include only a limited number of modifications that are specific, and that can be verified and validated.
(...효율적 검토를 위해, FDA는 PCCP에 구체적이고 검증 및 유효성 확인이 가능한 제한된 수의 modifications 만 포함할 것을 권장합니다.)
A. Goals of the Description of Modifications Section : FDA 는 modification protocol 이 PCCP 에 식별된 모든 수정을 지원하는 것이 아닌 일부 수정만을 지원
B. Content of the Description of Modifications Section : modifictions 는 상세 변경 목록, 구체적 변경 계획 등을 언급해야 함.
- 변경 사항을 설명할 시, 라벨링 변경을 참조하는 것이 도움이 될 수 있음. (Section VII.B 참고)
- PCCP는 구체적이고 검증 가능 해야함, modification은 Modification Protocol 내에서 성능 평가와 연계되어야 함 (Section VII.C Table1 참고)
- 계획된 변경이 자동으로 실행(*소프트웨어에 의한 자동실행) 인지, 사람의 결정에 의한 수동 실행 인지 명확하게 명시
- FDA는 자동으로 구현되는 PCCP 를 기존 장치와의 대응성, 안전성, 유효성에 대해 (요청 회사가) 적절히 검토하기를 권고
- 계획된 변경이 시장에 출시된 모든 장치에 균일 적용되는지, 장치에 따라 다르게 적용되는지 명확히 명시 (전역이 아닌 지역 적용일 경우 지역 적용의 정당성을 설명해야 함.)
C. Types of Modifications : 변경의 종류
- 양적 측정과 관련된 ML-DSF 성능 사양(performance specifications)의 변경 (*개인 의견: AUROC 가 아닐까 합니다)
- 장치 입력과 관련된 ML-DSF 수정 (*개인 의견: Input data 가 아닐까 합니다)
- 장치의 사용 및 성능과 관련된 제한적 수정 (예: 특정 하위 인구 군) (*개인 의견: 특정 환자군에 대한 AUROC 개선이 아닐까 합니다)
여기서 딥러닝 의료 인공지능을 만드는 기업들에게 중요한 부분이 언급됩니다.
Examples of modifications related to quantitative measures of ML-DSF performance specifications include improvements to analytical and clinical performance resulting from re-training the ML model based on new data within the intended use population from the same type and range of input signal.
ML-DSF 성능 사양의 정량적 측정 변경의 예시로는, 동일한 유형/범위의 입력 신호(input signal)에서 의도된 사용 집단 내의 새로운 데이터를 기반으로 ML 모델을 재학습함으로써 발생하는 분석 및 임상 성능의 개선이 있다.
즉 다시 말해, 분석 인공지능이 Input data 만 동일하다면.. 재학습한 새로운 인공지능 모델이 분석/임상 성능 개선이 있더라도 PCCP 로 포함시키겠다는 의미입니다.
개인적으로는 이건 이미 실질적으로 다른 제품이라 생각하지만, 1) 같은 의도된 사용 집단이라는 점, 2) 같은 INPUT 데이터라는 점 을 들어 같은 제품으로 인식해주겠다는 말로 보입니다.
또한, 데이터 학습과 관련된 추가 언급이 있습니다.
Modifications related to the device’s use and performance could include authorization of a device for a specific subset of a population within the originally indicated population based on re-raining on a larger data set for that subpopulation that was not previously available.
기기의 사용/성능과 관련된 변경은... 이전에 사용할 수 없었던 하위집단(subpopulation) 에 대한 더 큰 데이터셋에 대한 재학습을 기반으로... 본래 의도된 인구 집단 내부의 특정 하위 집단을 위한 기기를 포함합니다.
오역이 다분하지만, 해당 부분을 통해 짐작할 수 있는 부분은, intended use 와 관련된 학습 population dataset 이 더욱 큰 population dataset 을 학습할 경우에도 PCCP modification으로 고려중인 것으로 보입니다.
- PCCP 에 포함된 모든 변경은 장치의 의도된 사용 범위(device's intended use) 내를 유지해야함
- FDA는 PCCP 대상이 되는 모든 이익-위험, 해당 정책으로 인한 경험 등을 고려할 예정 (Appendix B 참고)
- Modification 절차
변경 절차(modification protocol) 는 PCCP 개발,검증,구현 문서화를 포함합니다. 여기서 QS(quality system)을 언급하며, PCCP 가 QS 규정에 따라 modification 검토, 승인 절차가 있어야 함을 언급하며(21 CFR 820.30 및 21 CFR 820.70), 변경 사항과 승인 내역을 제품표준서(device master record)에 문서화도록 요구함을 언급합니다.(21 CFR 820.181).
- 21 CFR 820.30 이란 품질시트템 CFR인 820 중에서, 설계 관리(Subpart C - Design Controls) 부분입니다. (DHF 전체를 말하는 CFR 입니다.)
- 21 CFR 820.70 이란 생산 및 공정 관리 CFR 입니다. (SOP 를 말하는 CFR 입니다.) 이 중, (i) 를 보시면 다음과 같습니다.
- 21 CFR 820.181 이란 제품표준서 CFR 입니다.
A. Goals of the Modification Protocol Section : modification protocol 이 장치의 안전성과 효과성을 보장해야 함을 강조합니다. 또한 modification protocol 은 개발, 검증, 배포에 대한 방법을 기술해야 함을 언급하며, modification protocl 이 description of modifications 문서와 연관되어 기술되어야 함을 언급합니다. modification protocol 의 목표는 다음과 같습니다.
- 개발, 검증, 배포 에 사용된 모든 방법과 데이터의 식별(identify)과 관련된 modifications
- 개발, 검증, 통계 분석, 특정 통과 기준과 관련된 modifications
- 제출되는 모든 '제안된 변경 사항'에 대한 정보는 제조업체에 의해 작성되어야 하며, 해당 정보들은 품질시스템에 따라 기록 보관 유지되어야 함
- Description of Modifications 에는 위험에 대한 상세한 영향 평가(impact assessment) 가 진행되어야 함.(변경과 관련된 식별된 위험. 업데이트 과정 및 사용자와의 커뮤니케이션 또한 포함)
이 목표들 중에 조금 재미있는 부분이 있습니다.
Be least burdensome for the manufacturer to develop and for FDA to review. This includes being traceable and specific to the modifications detailed in the Description of Modifications section and sufficiently comprehensive to support specific modifications.
(제조업체가 개발하는데 가장 덜 부담스러워야 하며, FDA가 검토하는 것에도 부담이 적어야 합니다. modifications 이 추적 가능하면서도 세부 정보를 충분히 포함하는 것을 말합니다.)
또한 해당 가이던스는 네 가지를 식별한다고 말합니다.
1. 데이터 관리 방법 (Data management practices)
2. 재학습 방법 (Re-training practices)
3. 성능 평가 절차 (Performance evalution)
4. 업데이트 절차 (Update procedures)
위 4 가지의 경우 아래 V. - B 에서 상세하게 설명합니다.
B. Content of the Modification Protocol Section : 변경 절차 내용
제조업체는 Modification Protocol 의 각 구성 요소에 대한 방법을 개요화해야 합니다. (각각의 Modification Protocol 구성 요소의 예시 요소는 부록 A 참고)
- 1. Data management practices (데이터 관리 방법)
- ML 에서의 학습 및 테스트 관련 데이터에 대한 변경 절차 입니다. 제조업체가 변경 사항을 구현하는 방법, 사용자에게 정보를 제공하는 방법, 사용자에게 훈련을 업데이트하는 방법, 실제 환경에서의 모니터링 수행 방법 등을 언급합니다.
- FDA 는 이와 관련하여 이 부분이 권고되는 이유를 다음과 같이 설명합니다.
1) 제조업체가 제안된 의도된 사용 집단을 완전하고 대표할 수 있는 교육 및 테스트 데이터를 확보
2) 식별 가능한 하위 인구들이 적절히 대표되도록, ML 모델을 편향을 최소화하기 위한 훈련 및 테스트 데이터셋 분리
3) 훈련 및 테스트 데이터가 과적합과 테스트 성능의 오인을 방지하기 위해 분리
4) 과거 데이터가 환자 인구와 치료 표준을 대표하도록 최신 데이터로 보충/대체
5) 참조 표준(reference standard) 이 최상의 가능한 절차를 대표
6) 데이터 관리 방법이 어떻게 잠재적 차별적 결과를 생성할 가능성을 감소
* 데이터 관리 방법은, FDA에게 ML-DSF 변경 사항이 사용 목적에 적합한 데이터를 기반으로 작성되어야 함
* 이 부분에는 ML-DSF 입력 데이터로 사용될 제품, 장치를 사용할 환자 인구 및 장치가 사용될 임상 시나리오 대한 정보가 포함
- 2. Re-training practices (재학습 방법)
- 재학습 시 모델 아키텍처의수정을 포함하는 경우도 언급하고 있습니다. (예: CNN에서 학습 하이퍼 파라미터 변경, 노드 수 변경, 레이어 수 변경) 이런 경우, modirication protocol 에서 재훈련 방법, 구체적인 아키텍처 변경에 대한 근거 및 정당성을 기술해야 합니다.
- FDA 는 이와 관련하여 이 부분이 권고되는 이유를 다음과 같이 설명합니다.
- 제조업체의 재훈련 방법에 대한 정보를 FDA 에 제공함으써, FDA 는 제안된 변경 사항이 적절한, 명확하게 정의된 방법에 따라 구현되는지 여부를 파악하고, 성능 평가 및 업데이트 절차가 수정 사항을 지원하는지를 결정할 수 있습니다.
- 3. Performance evaluation (성능 평가)
- FDA 는 위 변경 사항들에 대한 점검. 즉 해당 변경들에 대한 성능 요구사항을 PCCP 에서 제공하도록 요구합니다. 이 성능 평가는 각 개별 변경에 대한 검증 및 유효성 확인 계획이 포함됩니다. 예를 들어, 연구 설계, 성능 지표, 통계적 검정 등에 대한 정보가 있으며, 이 외의 테스트도 가능하다 언급합니다.
- FDA 는 이와 관련하여 이 부분이 권고되는 이유를 다음과 같이 설명합니다.
- 제조업체의 성능 평가 방법에 대한 정보는 FDA 에게 (의료기기의) 변경이 전체적인 성능에 미치는 영향을 평가하는데 사용됩니다. (여기서 적절한 연구 설계, 성능 지표, 통계적 검정이 사용될 것임을 확인하는데 도움을 준다, 라는 언급이 있는 것으로 보아 단순 AUC 검증이 아닌 각 변경 요소마다 (변경 전,후의 변화를) 검증할 객관적인 계획 및 방법을 요구하는 것으로 보입니다.)
- 추가적으로 modification protocol 을 진행하는 과정에서 성능 평가에 실패한 경우 실패 사항을 기록, 변경이 구현 및 배포되지 않음을 명시하라 표시되어 있습니다.
- 4. Update procedures (업데이트 절차)
- 업데이트 절차 부분에서는, 제품을 업데이트하여 변경 사항을 적용하고, 사용자에게 (업데이트 된) 장치를 안전하게 사용할 수 있도록 관련 정보를 전달해야 함을 강조합니다. 예를 들어, 제조업체가 장치를 변경/구현하는 방법. 사용자에게 투명성을 제공하는 방법. (업데이트를 진행한) 사용자 교육 및 모니터링, 의도하지 않은 작동에 대한 통제 등이 있습니다.
- FDA 는 이와 관련하여 이 부분이 권고되는 이유를 다음과 같이 설명합니다.
- 1) 변경 구현으로 인한 위험을 업데이트 절차가 어떻게 (위험을) 완화하는지
- 2) 업데이트 통지 방법
- 3) 업데이트 이후 장치가 안정적 작동/유지 방법
- 4) 모든 이해관계자가 장치 기능/성능에 대한 최신 정보 획득
- 5) FDA 가 업데이트 절차 자체와 관련된 잠재 위험 완화 이해
C. Traceability Between the Description of Modifications Section and the Modification Protocol Section
변경 설명 섹션 - 변경 절차 섹션 간 추적성 부분입니다. 변경 설명(Description of Modifications) 과 변경 절차(Modification Protocol) 를 명확히 분리할 것을 언급하는데, 이를 추적성 테이블(traceability table)로 기록하는 방법을 예시로 다음과 같이 제공합니다.

- 영향 평가
가이던스 마지막 항목인 VI. Impact Assessment 입니다. (물론 이후 Appendix 가 더 있습니다) 모든 의료기기들이 비슷하겠으나, SW 는 특히 업데이트 이후 어떤 버그가 발생할지, 사용자가 어떤 변화를 겪는지 등의 업데이트 이후 여러 결과가 발생합니다. 따라서 이러한 업데이트(변경) 들은 이후 어떤 영향이 나타날지 impact 를 평가해야 합니다.
FDA는 본 Impact assessment 로 다음과 같은 영향 평가 내용을 문서에 포함하길 권고합니다.
1) 각 변경이 적용된 장치의 버전을 아무 수정도 적용되지 않은 버전과 비교 (업데이트 vs 비 업데이트)
2) (사회적 피해 위험을 포함한) 각 개별 변경의 이득/위험 논의
3) 변경 절차에서 제안된 사항이 기기의 안전성 / 유효성을 합리적으로 보장
4) 하나의 변경이 다른 변경 실행에 어떤 영향을 미치는지 논의
5) 모든 변경의 종합적인 영향 논의
-> 이를 통해 제안된 변경들의 조합이 추가적인, 미완화된 위험을 도입할 가능성이 적음 + 및 기기의 안전성 및 유효성이 유지됨을 입증
* 만일 ML-DSF 가 다중 기능 장치(Multiple Function Device Products) 일 경우, 해당 가이던스를 참고하여 문서화 할 것을 권고합니다.
해당 가이던의 Appendix 는 A와 B가 있습니다.
해당 Appendix 의 경우 상세하고 세부적인 가이던스를 제공함으로 해당 부분은 다음 post 에서 다루도록 하겠습니다.
'인허가 > FDA' 카테고리의 다른 글
| Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program - FDA 가이던스에 대하여 part 1 (0) | 2023.07.28 |
|---|